 全国服务热线
全国服务热线153-3803-1685

第三章 芳香族聚酰胺纤维
第一节 概 述
自从美国杜邦公司科学家 Carothers1935年发明脂肪族聚酰胺,即聚己二酰己二胺(尼龙 66)纤维以来,聚酰胺纤维作为工业用纤维,发挥了很大作用。一直到 60年代,有好多种新聚酰胺产品问世,其中最重要的发现是杜邦公司 1962年发表的聚间苯二甲酰间苯二胺纤维(当时称为 HT-1),以后正式商品的名称为 Nomex(R。到 1966年又发表更令人注目的高性能聚对苯二甲酰对苯二胺纤维(当时称为 FiberB),其正式的商品名为 Kevlar(R。为了区别于脂肪族聚酰胺的通称,1974年,美国政府通商委员会把芳香族聚酰胺通称定名为 Aramid,泛指酰胺基因直接与两个苯环基团连接而成的线形高分子,用它制造的纤维就是芳香族聚酰胺纤维(Aramid纤维)。在我国又称为芳纶,间位Aramid纤维称做芳纶 1313,对位 Aramid纤维称做芳纶 1414,其数字部分表示高分子链节中酰胺键和亚胺键与苯环上的碳原子相连接的位置(参见芳纶的分子结构式)。芳纶和普通聚酰胺纤维相比,性质和用途有很大区别。芳纶 1313耐高温性好,不会熔融;芳纶 1414强度高、模量高又耐高温。这类纤维主要用在工业技术性能上有特殊要求的产品中,因此最初就称为特种合成纤维。同时,以芳纶为代表的特种纤维,无论对高分子的合成技术还是纤维成形工艺方面,都反映着高科技的水平,工艺步骤有时非常复杂,物料的管理十分严格,在基础理论上也引入了许多新的概念,例如刚性链大分子结构、高分子液晶理论、干湿法纺丝成形技术等,有许多研究工作目前还在深入下去。芳纶 1313和芳纶 1414纤维,前者作为耐热纤维,后者作为高强度高模量纤维都得到了很大的发展。当时同期研究开发芳香族聚酰胺的还有其它一些纤维公司,大多数公司都碰到如高分子的相对分子质量做不高、溶解方面选不出合适的溶剂等等困难而中止了研究。其中美国 Monsanto公司开展了改性芳纶的研究工作,称为 X-500,由对氨基苯酰肼(PABH)和对苯二甲酰氯,在二甲基乙酰胺(DMAc)氯化锂的质量分数为 5%
的溶剂体系中,低温溶液缩聚反应,反应浆液用碳酸锂中和,并调配成聚合物的质量分数为 7 7%的纺丝原液,是各向同性溶液,经过湿法纺丝拉伸后得到纤维,但是纤维的综合性能没有 Kevlar(R纤维好,以后放弃了工业化研究,这是最早对位芳纶的化学改性尝试。Kevlar(R纤维的问世,代表着合成纤维向高强度、高模量和耐高温的高性能化方向达到了一个新的里程碑,成为高科技纤维工业的先驱。
一、芳香族聚酰胺纤维发展简史
芳纶的开发是在脂肪族聚酰胺研究的基础上进行的,60年代 H Mark在用苯环基团替代脂肪基团(—CH2—)时,发现大分子链变得坚韧、熔点上升、吸湿性降低等现象,因此进行了系统的研究,其中芳纶 1313缩聚浆液可以作为纺丝原液,容易制成纤维。同时期,美国政府正在实施宇宙太空计划,迫切需要耐热纤维材料,使得耐热纤维的研究显得更加重要。杜邦公司 1962年发表的间位 Aramid纤维(HT-1),1966年完成工业化,于 1967年正式以 Nomex(R的商品名上市,是耐高温性能极好的纤维。目前该公司在美国本土的生产能力为 1 5万 t/a,在西班牙的独资企业生产规模1993年为 5000t/a。在太空竞争中,前苏联也开发了相类似的芳香族高分子纤维,建造了工业生产装置。
在芳香族聚酰胺的研究中,从分子构造的理论与实验可以推测,对位连接的芳酰有利于提高强度,又因它的玻璃化温度高,熔点高于热分解温度,常规有机溶剂中不溶解,用普通的纺丝方法无法制得纤维。1966年杜邦公司科学家 S L Kwolek发现在某些条件下,对位芳纶可溶解在浓硫酸中,达到临界浓度时,形成高分子液晶溶液,发明了液晶纺丝技术,纺出的纤维强度非常高。1971年杜邦公司完成试生产设备(100~200t/a),1972年正式公开的商品名称为 Kevlar(R。1996年美国本土的生产能力 2 1万 t/a,在英国北爱尔兰的工厂 5000t/a,在日本与东丽公司合资的东海工厂 2500t/a,近期将扩大一倍,因此杜邦公司的 Kevlar(R总生产能力超过 3万 t/a。在欧洲,1975年开始研究对位芳纶,1995年生产能力为 5000t/a。
在日本,芳香族聚酰胺纤维也得到开发,1972年帝人公司发表了芳纶 1313同类产
品,商品名称为 TeijinComex(R,目前年生产能力为 2300t。1987年发表芳香族共聚酰胺纤维,化学结构不同于 Kevlar(R,但生产规模只有 750t/a。
俄罗斯的芳香族聚酰胺纤维,主要是含杂环类的共聚芳纶,机械性能和耐热性方面
比较好,由于没有形成规模效应,因而产量少,成本高,目前只应用于航天器材和高性能复合材料等领域。
二、芳香族聚酰胺的结构与性能
芳香族聚酰胺最简单的化学结构聚对苯甲酰胺(PBA), -AB-型缩聚物,而 PPTA
是 -AA-BB-型缩聚物,酰胺键之间可以连接多种苯环基团,形成各式各样的刚性链结构,其共同的特征是链节单位在大分子链上呈同轴或平行的伸展结构,表 3-1列举典型芳香族聚酰胺纤维的实例。

MPIA分子结构中酰胺键和间位苯环连接,间位苯环上的共价键内旋转位能低,可
旋转角度大,因此 MPIA大分子是柔性链结构,所以在力学性能上接近普通的柔性链纤
维,但苯环基团含量高,耐热性能就大于脂肪族纤维。而 PPTA纤维是对位连接的苯酰
胺,酰胺键与苯环基团形成共轭结构,内旋位能相当高,成为刚性链大分子结构,分子
排列规整,因此分子结晶和取向极高,所以纤维的强度和模量相当高。这种结构上的差异,使间位芳纶和对位芳纶在力学性能上区别特别大,因而在应用上也有很大的不同
(在后面章节再详细介绍)。
第二节 聚对苯二甲酰对苯二胺纤维
一、单体和合成
和通常的聚酰胺一样,PPTA用缩聚的方法合成。但由于熔融温度高于聚合物的分解温度,不能用熔融缩聚的方法,只能用界面缩聚、溶液缩聚和乳液聚合的方法,作为研究还有固相缩聚和气相聚合的方法。工业生产上常用低温溶液缩聚和界面缩聚的方法,由芳香族二胺与芳香族二酰氯,在酰胺型溶剂体系中反应制备聚合物,其反应式如下所示。
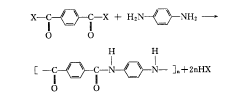
其反应是 Schotten-Baumann型反应,如下所示。
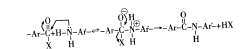
因此反应与 -X基团的性质有关,能形成酰胺键的 -X有下列几种:
—COCL X:卤素
—COOR X:OR(R烷基,芳基)
—COOH X:羟基
这些化合物中,当—X为卤素—Cl时,作为反应单体活性最高,因此芳香族二酰氯是首
选单体,对苯二甲酰氯用得最多。
1 界面缩聚
界面缩聚法是两种单体分别溶解在两个不相混溶的溶剂中,即把对苯二甲酰氯溶解
于与水不相溶的有机溶剂中,对苯二胺溶解在水相中,当两种溶液相互混合时,在相的界面就发生缩聚反应生成聚合物,因为聚合物不溶解在两个溶剂里,因此以沉淀形式析出,在界面缩聚反应时,单体及聚合物的浓度分布如图 7-3-1所示。
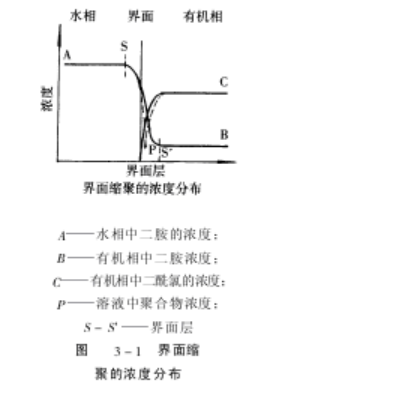
图中的界面层是发生缩聚反应的区域,水相中高浓度的二胺向有机相扩散,扩散速
率决定着缩聚的速度,在 P点产生聚合物的沉淀,其浓度最小。在界面缩聚中,反应发生在界面层里,因此界面的产生、更新及二胺的扩散速率等反应条件起了重要作用,对聚合物的相对分子质量影响很大。
2 低温溶液缩聚
采用反应活性大的单体如二酰氯和二胺,在非质子极性溶剂如 DMAc、N-甲基吡咯烷酮(NMP)、六甲基磷酰三胺(HMPA)等酰胺型溶剂,在温和的条件下进行缩聚反应的方法称为低温溶液缩聚法。适合于反应活性大,热敏性高的单体,在室温以下进行反应,可以避免副反应发生,得到高的相对分子质量的聚合物,这个方法符合芳香族聚酰胺合成的要求,下面将详细介绍。
3 固相缩聚
缩聚过程物料以固体形式进行化学反应,得到高相对分子质量的方法称为固相缩
聚,一般先把单体缩聚成低相对分子质量的聚合物,再进一步在高真空下,或在惰性气氛保护下加热到熔点附近,发生链增长的缩聚反应,这个方法在芳香族聚酰胺的合成中用得极少。在芳香族聚酯中,初生纤维的长时间热处理过程,有点类似固相缩聚的方法。
工业生产上使用低温溶液缩聚法制备 PPTA聚合物。对苯二甲酰氯(TCl)和对苯二
胺(PPD)为单体,NMP-CaCl2即酰胺 -盐溶剂体系为缩聚溶剂,选择合理的缩聚反应工艺是得到高相对分子质量、具有伸展链结构 PPTA的关键。
1)PPTA缩聚工艺流程
对苯二胺与对苯二甲酰氯的低温溶液反应工艺流程见图 3-2所示,它和通常的
缩聚反应一样,遵循 Flory有关缩聚的公式:
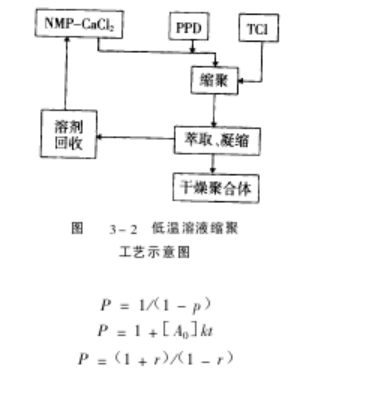
可见聚合度 P与反应程度 p,单体摩尔配比 r=[A0] /[B0],反应时间 t密切相关,其中[A0]、[B0]分别是两个单体的初始摩尔浓度。工业化生产时,由于设定 p, t条件相同,因此两个单体的摩尔配比 r对聚合物的相对分子质量影响就特别明显。
2)单体纯度
单体的纯度和缩聚反应时的操作条件如计量精度、溶解完善与否等机械因素,都会影响两个单体的摩尔配比 r。从上述的公式可以看出,只有在等摩尔配比时,才会得到高相对分子质量的聚合体。工业化生产时,由于机械设备已经定型,操作高度自动化,所以机械因素的影响可以排除,而单体的纯度就十分重要了。二个单体中对苯二甲酰氯性质特别活泼,存储和使用时稍有不当,纯度就有变化,而这种变化不容易发现,因此使摩尔配比发生偏差直接影响相对分子质量,它的大小可用对数比浓粘度$inh表示,相互关系如表 3-2所示。表中数据显示,对苯二甲酰氯的纯度稍有变化,对聚合物的相对分子质量影响相当大,要获得高$inh,纯度就要 99 9%以上。同样另一个单体对苯二胺也要求纯度高,只有这样才能精密计量,如果对苯二胺纯度不高,还会使聚合物的颜色变深。
表 3-2 对苯二甲酰氯纯度与聚合物的$inh关系

3)溶剂体系
低温溶液缩聚反应,使用溶剂的数量大,对苯二甲酰氯反应活性大,热力学上的惰性溶剂才可使用,同时为了获得高相对分子质量,要尽量增加聚合物在溶液中的溶解度,以便提高反应程度,所以只有酰胺型溶剂体系才符合上述条件,它们的性质列于表3-3。
表 3-3 缩聚溶剂的性质
杜邦公司的专利曾报道,制造高相对分子质量 PPTA常使用酰胺类混合溶剂,比单
一溶剂效果更好,如 HMPA-NMP,HMPA-DMAc等混合溶剂,其中因为 HMPA吸收反
应副产物盐酸好,两个溶剂产生协同效应,对 PPTA的溶解性也高,所以普遍使用。但
是 1975年以后,人们发现 HMPA有致癌作用,且回收上有难度,从那时起放弃使用
HMPA。工业生产上改用酰胺 -盐溶剂体系,单独采用 NMP溶剂,它是比较安全的,但性能上比 HMPA混合溶剂稍差些,可通过加入碱或碱土金属盐,产生溶剂分子与金属阳
离子的多级缔合作用,例如组成如 NMP-CaCl2,NMPLiCl等体系,溶剂中存在的金属阳
离子,将增加体系溶剂化作用,加强溶剂体系与 PPTA之间亲合,增加 PPTA的溶解性,
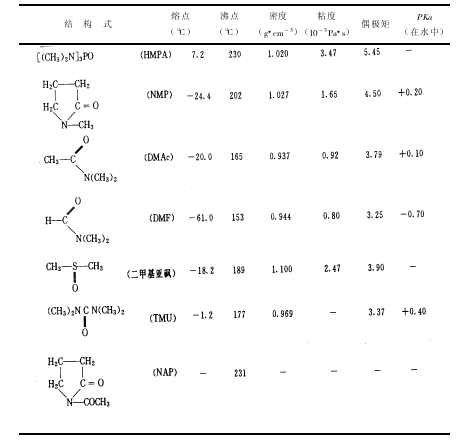
促进缩聚反应的程度。在 NMP-CaCl2体系中,CaCl2的含量对聚合物的粘度$inh的关系
见图 3-3所示,当 CaCl2质量分数达到一定值时,聚合物的相对分子质量最高。不同的溶剂体系对 PPTA的合成反应影响也是不同的,因为溶剂化作用不同,吸收
或排除缩聚过程中放出的副产物如盐酸的程度不同,还有副反应的控制也不一样,所以
选择溶剂体系对合成 PPTA至关重要。
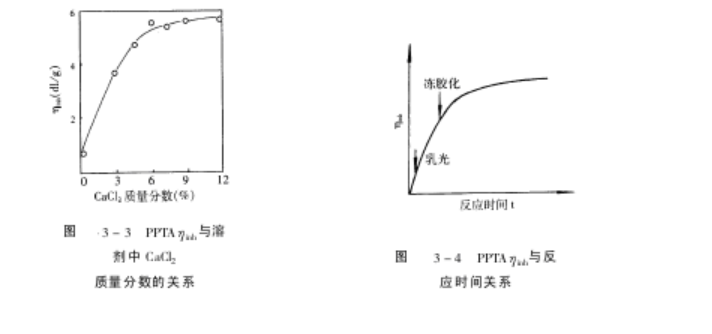
4)反应时间
缩聚反应是逐步聚合过程,缩聚公式表明相对分子质量与反应时间有关,PPTA合成时,其对数比浓粘度$inh随反应时间的变化如图 3-4所示。反应速度极快,反应开始 30~90s,反应体系产生乳光效应,几分钟后产生爬杆现象,随即发生冻胶化,对初生冻胶体加以强力剪切作用,聚合物的$inh会增加,反应后期$inh不再增加。
5)反应温度
对苯二胺与对苯二甲酰氯的缩聚反应速度极快,又是一个放热反应,根据反应热效
应的测定,缩聚反应热为 -199kJ/mol,反应体系总的反应热大致在 -245! 265kJ/mol,因此控制剧烈放热下的反应温度相当重要。反应温度过高,将会增加副反应和聚合物的降解,选择低的反应初始温度,有利于得到高相对分子质量的聚合物。按照制备高强高模纤维的要求,聚合物的ηmin为 5 .5---6 .5dl/g时,才适合进行纺丝。
上面讨论了 PPTA合成的工艺条件,工业化生产聚合物希望缩聚过程能够连续进行自动化控制,以降低成本、稳定纤维的质量。针对缩聚反应单体严格的摩尔分数,随着相对分子质量增加、反应体系迅速冻胶化、以及大量反应热下反应温度的控制等等,工业生产已经设计了特殊的反应混料器,使对苯二胺的酰胺 -盐溶液和熔融的对苯二甲酰氯,连续迅速地混合反应,用计量泵精确控制单体的摩尔分数,物料在混合室里停留极短时间,立即进入双螺杆反应器,在高剪切下完成缩聚反应,温度也控制在较低的范围内,最后高相对分子质量的聚合物粉碎以屑粒形式排出,缩聚溶剂回收利用,聚合物干燥后供给纺丝工序。PPTA在浓硫酸溶液中,特性粘度[η]与相对分子质量 Mr有如下的关系
[η]=7. 9 ×10-5Mr1 .06
通常特性粘度值在 4以上时,PPTA相对分子质量大于 27000。
PPTA其它的合成方法有气相缩聚法、固相缩聚法等。还有不用对苯二甲酰氯,直接采用对苯二甲酸和对苯二胺,在吡啶及苯基亚磷酸盐催化剂作用下发生直接缩聚。但是这些方法聚合物的相对分子质量目前还做不高,尚在继续研究中。一些新的合成方法,如络合催化的方法等也有文章报道。
PPTA由于规整的刚性大分子结构和分子间氢键的作用,溶解性能不理想,缩聚溶剂和纺丝溶剂不一样,带来诸多不便。PPTA纤维开发成功后,为了改进性能,对共聚PPTA进行开发,其中代表性的芳香族共聚酰胺是日本的 DPEPPTA,由 3,4-二氨基苯醚(3,4-ODA)、对二苯胺与对苯二甲酰氯在 NMP酰胺型溶剂中低温溶液缩聚,反应方程式如下所示:
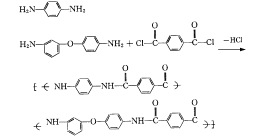
得到聚合物溶液用氧化钙中和,调整溶液中聚合物的质量分数为 6%就成为纺丝原液,这个溶液呈现各向同性性质,因为大分子链上引进了醚键和间位苯环基团,提高了聚合物的溶解性,反应产物能够溶解在缩聚溶剂中,简化了溶剂回收工艺。
二、纤维成形
PPTA纤维成形技术是典型的由刚性链聚合物形成液晶性纺丝溶液,采用杜邦公司发明的干喷湿纺的液晶纺丝方法,制取高强度高模量纤维。和传统的熔融纺丝、湿法纺丝及干法纺丝的方法相比,引进了新的概念和理论基础。在工艺技术上,如聚合物在浓硫酸中溶解时,溶液的粘度开始很高,聚合物质量分数达到临界值后,溶液粘度逐步下降,呈现液晶的特征,溶液体系只有在高于转变温度时才成为液态状,因此纺丝溶液的制备要碰到高粘度的搅拌和脱泡的技术困难,还要克服聚合物在高温溶解条件下的降解问题等等。这些技术难题正是利用了高分子溶液的液晶理论和干喷湿纺的纺丝工艺技术,得到完美的解决使高性能 PPTA纤维实现了工业化生产。
1 纺丝原液的制备
刚性链的 PPTA在大多数有机溶剂中不溶解,也不熔融,只在少数强酸性溶剂中例如浓硫酸、氯磺酸和氟代醋酸等强酸溶剂里才溶解成适宜纺丝的浓溶液。表 3-4是几种强酸的物理性质。从工业化生产上考虑,选择浓硫酸做 PPTA的溶剂比较适合。硫酸酸性强,溶解性能适中,挥发性低,回收工艺成熟,比较经济,和其它强酸比优点较多。硫酸分子与刚性链 PPTA分子可能以下列形式化合:

沿着大分子链发生质子化作用,促进了溶解进程,测定聚合物在溶剂中溶解后的特性粘度,可以评价溶解情况。PPTA分别溶于 100%和 96%的硫酸中,其特性粘度值之比为 10~15,说明在 100%的硫酸中溶解性比较好,特性粘度值比较高。从上面的反应式也看出,自由负离子 HSOZ4 的浓度,将影响溶解性能,如果体系中存在水分,会使自由负离子浓度增加,则体系的溶解性下降,所以 96%的硫酸溶解性比 100%的硫酸要差。
研究表明浓度为 99% ! 100%的硫酸,对 PPTA的溶解性最好。随着 PPTA的浓度增加,溶液粘度上升,当到达临界浓度以后,粘度又开始下降,因为刚性棒状大分子,在低浓度时,溶液是各向同性体,所以粘度随浓度而上升。当溶液浓度过了临界浓度以后,刚性分子聚集形成液晶微区(Domain),在微区中大分子呈平行排列状,形成向列型液晶态(NematicLiquidCrystal),但各个微区之间的排列,呈无规状态。当溶液受到一点外力场作用时,这些微区很容易沿着受力方向取向,因此粘度又开始下降,其关系如图 3-5所示。随着温度上升,曲线向右移动,表明临界浓度值向高浓度一侧移动,有利于高浓度纺丝浆液的生成,有利于纺丝纤维的强度变大。
根据 Flory理论,溶液中刚性链大分子的临界值浓度( Vc)与大分子轴比( X)的
关系,近似地可用下式表示
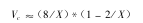
刚性大分子的轴比与其相对分子质量成正比,因此 Vc值与相对分子质量有密切关系,从 Vc值可以粗略地估算大分子链的长度和形成液晶的情况。
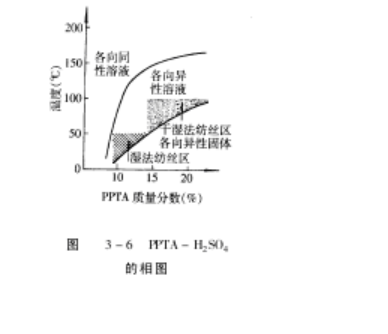
对于溶液纺丝来说,一般希望聚合物相对分子质量尽量的大些,纺丝原液的浓度应该高些,而其粘度要低些以利于成形加工。从 PPTA-H2SO3纺丝原液的相图,(图 3-6)来看,在聚合物质量分数为 18% ~ 22%的范围里,溶液的粘度在 90 时,处于可
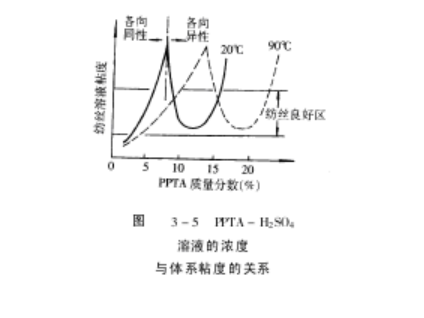
纺性良好的低粘度区(参看图 3-5),低于 80 时体系呈固态,所以相图和相转变
的研究,对于纺丝原液的制备十分重要
溶液纺丝时要求凝固浴的温度低一些,有利于大分子取向状态的保留和凝固期间纤维内部孔洞的减少,低温凝固浴的温度为 0 ~5 ,而纺丝原液是高温状态,因此喷丝头不能浸入凝固浴中,而干喷湿纺过程,允许高温原液和低温凝固浴的独立控制。

微信公众号
